
Figure 1. Taken from Ref. [1].
The Angew. Chem. paper I am sharing with you this time is
impressive, it is on the chemistry-biology interface. [1] It is about bio-conjugation
, which is applying a chemical reaction to a biological macromolecule. Let’s
deal with both fields in detail, one by one.
Bioconjugation is useful, because we can start to attach unnatural
or unconventional chemical substrates onto biological molecules, such as
proteins, complex carbohydrate architectures or nucleic acid-based compounds.
While these methodologies have found great uses in synthetic biology, the
design of the reactions is often challenging. Unlike conventional organic
substrates, the reactants we are dealing with here are biological molecules,
which are sensitive to high temperature, extreme pH, and possess loads of
stereogenic centers. Conventional wisdom shows us that we can lower the
activation energy barrier of a reaction (i.e. increasing the reaction rate) by
increasing the reaction temperature. However, this does not help in the case of
biological molecules, as the high temperature, or a sudden perturbation in pH,
can lead to negative results, such as the denaturation of proteins or possible
racemizations. So, the best methods for bioconjugation should work optimally at
physiological temperature (37oC) , narrow pH range, and also work
well in an aqueous media.
How can we incorporate unnatural chemical functionalities
into a supposedly natural biomolecule? Molecular biology methods, of course! In
this case, a protein was expressed with an unnatural amino acid (UAA), with a
carbon-carbon triple bond (alkyne) on it. The alkyne can act as a ‘handle’ for
other reactions, and in the past, click chemistry and Sonogashira coupling has
already been successfully employed.

Figure 2. The Glaser-Hay coupling reaction.
The reaction in focus is known as the Glaser-Hay coupling
reaction (Figure 2). It is a copper-catalyzed coupling reaction between 2 carbon-carbon
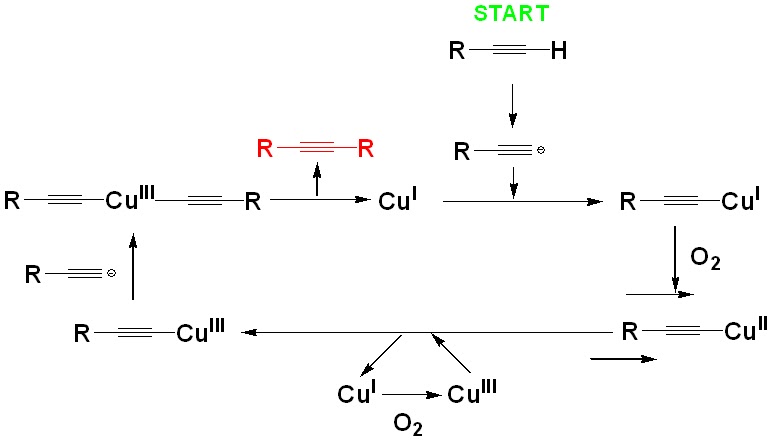
triple bonds. Previous studies have concluded that the reaction probably
involved a dimeric copper acetylide intermediate, and the mechanism involved an
oxidation of the copper species, from +1
to +2 and then +3, and finally back to +1, continuing the catalytic
cycle (Figure 3). Traditionally, this was a homo-coupling reaction, which meant you had 2
identical coupling partners to get the dimeric, symmetrical product. This
unfortunately hinders its potential applications. Because what we want is a
CROSS-coupling (hetero-coupling) reaction, in which you have 2 different
coupling partners, A and B, to form an A-B type product (rather than an A-A or
B-B type product). How about adding 2 different substrates to the Glaser-Hay
system? Nope, it does not work well, as we almost alyways have a statistical
amount of homo-coupling products, and that hinders purifications too. There is
good news, though, as recently novel systems have been developed to lead
predominately to hetero-coupled partners. For example, Lei and Agrofoglio
independently developed co-catalyzed system involving a Ni (II) / Cu (I)
system, and these methods work well for a variety of alkynic substrates. On the
other hand, if one of the coupling partners is attached to a solid support,
heterocouplings also predominate. Glaser-Hay reactions work well in organic
solvents, yet it is rather unprecedented to work in an aqueous media. This
article shows us that possibility.

Figure 4. The substrate for the coupling reaction. Taken from Ref. [1].
The researchers started with some model reactions on small
molecules before embarking on biomolecules (Figure 4, a). With phenylacetylene and propargyl
alcohol respectively, they could achieve respectively homo-coupled products in
an aqueous solution with CuI / TMEDA as the catalytic system. They had to
premix these 2 reagents to establish the copper complex before it could be
bound to an alkyne substrate and start catalysis (Figure 5).

Figure 5. The first stage of the catalytic cycle. Taken from Ref. [1].
To achieve unconventional biological products for their methodolgy,
the researchers installed an unconventional amino acid, ‘propargyl
phenylalanine’, at position 151 of the green fluorescent protein substrates
they used, which was known as pPrF-GFP (Figure 4, b). They coupled this substrate with ‘AlexaFluor
488 modified alkyne’, and succeeded in making the hetero-coupled product (Figure 6, 7). They
have also explored the substrate scope of their method. For solid-supported
reactions, they have achieved similar yields with a propargyl or a hexyl-derived
alkyne, and also in other types of biologically-related substrates (Figure 8). The results
were easily visualized, as a successful coupling reaction would lead to an
observable fluorescent signal, or by SDS-PAGE analysis (Figure 7).

Figure 6. Glaser-Hay bioconjugation. Taken from Ref. [1].

Figure 7. The results of the Glaser-Hay bioconjugations as illustrated
by a normal SDS-PAGE gel and a fluorescent version. In diagram A, we can see
that the unconventional amino acid, pPrF, has been successfully incorporated
into the GFP (Lane 2), because there is no GFP spot in Lane 3, when pPrF is
absent. In diagram B, we can observe that the coupling reaction has been
successful carried out because in Lane 2 and 4, which corresponds to 2
different temperatures, and with both copper catalyst and pPrF-GFP present,
this substrate is coupled to the fluorophore ‘Alexa’. Therefore, fluorescene
signals are evident in those lanes, signifying successful C-C bond formation.
This is not the case for the other control experiments (Lane 3, 5, 6, 7), where
one or both of the aforementioned components are absent. When the researches
use a wild-type GFP – that means one that has no pPrF incorporated, of course
no coupling occurs.

Figure 8. Other substrates. Taken from Ref. [1].
The researchers have discovered a possible unproductive side
reaction - a Cu (I) -catalyzed oxidative degradation. Another interesting
observation was that the reaction rates were comparable at a low (4 oC)
and high temperature (37 oC). They attributed this to a protein degradation at the higher temperature,
inhibiting reaction progress.
Some personal opinions
This paper is absolutely smashing, and I hope there will be
further developments on the project. In the paper, the researchers have
employed 2 different alkynic substrates, a propargyl and a hexyl one. I wonder
if there will be any different, in terms of reaction rates, as the chemical
environment next to the 2 respective triple bonds are surely distinctive – in
the propargyl case, the triple bond and the oxygen is only separated by a CH2
group, while in the hexyl alcohol case, the oxygen and the triple bond were
separated by a much longer spacer. A further question would be, can we use a
more hindered propargyl-type secondary ether, as we may even generate a
possible stereogenic center close to the biological site (Figure 9)? The ‘double
conjugated alkyne’ should serve as a nice handle for further reactions to occur
– how about a thiol-yne coupling or a nucleophilic addition of ROH onto the
terminal alkyne?

Figure 9. A potential asymmetric Glaser-Hay coupling reaction?
The second question - is it possible to use a Cu (II) salt,
such as CuSO4 , instead to effect the coupling reaction? Obviously,
this would not have the same mechanistic similarity like the Glaser-Hay type
reaction, yet homocoupling involving Cu (II) salts do exist, e.g. Cu(OAc)2
for Eglinton reaction. If this is OK, then it will increase the versatility of
the reaction as the Cu (II) salts are readily accessible.
It would be really exciting if this research topic can be
developed much further!
by Ed Law
28/6/2015
Reference:
1. Development and Optimization of Glaser–Hay
Bioconjugations
J. S. Lampkowski, J. K. Villa, T. S. Young, and D. D.
Angew. Chem. Int. Ed., 2015, asap.
DOI: 10.1002/anie.201502676











